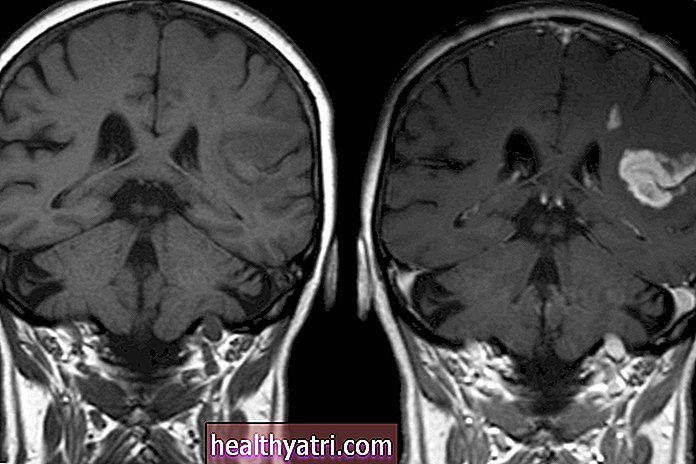
De ziekte van Huntington is een zeldzame aandoening die ongeveer 1,22 op de 100.000 mensen in de Verenigde Staten treft. De ziekte komt voor in families en wordt altijd veroorzaakt door een erfelijk gen.
Het genetische defect dat verband houdt met de ziekte van Huntington veroorzaakt degeneratie van zenuwcellen in bepaalde delen van de hersenen die beweging en denken beheersen. Na verloop van tijd leidt een geleidelijk progressieve achteruitgang van de hersenen tot de kenmerkende symptomen van de ziekte van Huntington.
Yuichiro Chino / Getty-afbeeldingen
Overervingspatroon
Erfelijkheid is de enige bekende oorzaak van de ziekte van Huntington. Het wordt overgeërfd in een autosomaal dominant patroon. Als een persoon het gen erft dat de ziekte van Huntington veroorzaakt, dan 'domineert' het ziekteverwekkende gen de andere, normale niet-ziekteverwekkende versie van het gen, en zal de persoon de ziekte zeker ontwikkelen.
Iedereen die de ziekte heeft, moet ten minste één kopie van het ziekteverwekkende gen hebben. Als een persoon het gen draagt dat de ziekte van Huntington veroorzaakt, heeft elk van zijn nakomelingen een kans van 50% om het defecte gen te erven.
Aangezien de ziekte gewoonlijk tussen de 30 en 50 begint, zouden veel mensen met de ziekte geen symptomen hebben ontwikkeld tegen de tijd dat ze kinderen krijgen.
In een gezin waarin één ouder het gen heeft, zou ongeveer de helft van de broers en zussen het gen erven dat de ziekte van Huntington veroorzaakt, en daarom de ziekte ontwikkelen.
Nakomelingen van een persoon met de ziekte van Huntington hebben ook een kans van 50% om het gen niet te erven - en zouden de ziekte dus niet ontwikkelenofde ziekte doorgeven aan hun eigen kinderen.
Begrijpen hoe genetische aandoeningen worden overgeërfd
Genetica
Het HTT-gen is het gen dat de ziekte van Huntington veroorzaakt en bevindt zich op chromosoom vier. Elke persoon erft twee exemplaren van chromosoom vier, één van hun biologische vader en één van hun biologische moeder.
Het genetische defect dat de ziekte van Huntington veroorzaakt, wordt beschreven als een CAG-herhaling in het HTT-gen. Dit is een mutatie (wijziging van de normale volgorde) in het deoxyribonucleïnezuur (DNA) -molecuul.
De mutatie bestaat uit een herhaald patroon van cytosine, adenine en guanine, nucleotiden in het DNA-molecuul die coderen voor de productie van lichaamskenmerken.
De meerderheid van de patiënten met de ziekte van Huntington hebben 40 tot 50 CAG-herhalingen met een normaal aantal herhalingen van minder dan 28. Deze extra nucleotiden veranderen de instructies van het HTT-gen met als gevolg de productie van een abnormale of mutant voor het huntingtine-eiwit. Iemand met de ziekte van Huntington hoeft niet per se hetzelfde exacte aantal CAG-herhalingen te hebben als de ouder van wie hij de ziekte heeft geërfd.
Juveniele ziekte van Huntington
Er is ook een juveniele vorm van de ziekte van Huntington die begint tijdens de kindertijd of jongvolwassenheid en sneller voortschrijdt dan de volwassen vorm van de ziekte, waarbij op jongere leeftijd ernstiger en sneller progressieve effecten optreden.
De juveniele vorm, die hetzelfde erfelijke autosomaal dominante patroon volgt als de volwassen vorm, wordt geassocieerd met een hoger aantal CAG-herhalingen dan de volwassen vorm. Mensen met de juveniele ziekte van Huntington hebben gemiddeld ongeveer 60 CAG-herhalingen in het HTT-gen.
Eiwitten herstellen
Naast de CAG-herhaalmutatie van het HTT-gen, hebben mensen met de ziekte van Huntington ook gendefecten in genen die coderen voor eiwitten die helpen bij het repareren van DNA.
Deze eiwitten helpen de normale structuur van het DNA te behouden en kunnen CAG-herhaalmutaties helpen voorkomen. Er zijn aanwijzingen dat het hebben van meer defecten in deze reparatiegenen ook zou kunnen leiden tot meer CAG-herhalingen en een eerder begin van de aandoening.
Hersenen veranderingen
Studies tonen aan dat mensen met de ziekte van Huntington afwijkingen hebben van de caudate en putamen-gebieden van de hersenen die normaal gesproken worden geassocieerd met denken, geheugen, gedrag en motorische controle.Veranderde functie van de neurotransmitters, vooral dopamine, in deze gebieden kan spelen een rol bij de ziekte van Huntington.
Deze veranderingen omvatten atrofie (krimpen), evenals afzettingen van materiaal, zoals cholesterylesters (CE), een soort vetmolecuul.
De ziekte van Huntington wordt geassocieerd met inflammatoire vernietiging van hersencellen die voorheen functioneel en gezond waren. Aangenomen wordt dat het defecte huntingtine-eiwit een rol speelt bij het ontstaan van de ziekte. De functie van dit eiwit is niet met zekerheid bekend, maar het kan een rol spelen bij het beschermen van hersencellen tegen gifstoffen.
Bij de ziekte van Huntington treedt een proces op dat wordt beschreven als autofagie wanneer cellen worden vernietigd en vervolgens degenereren. Er wordt voorgesteld dat de ziekte kan optreden als gevolg van schade veroorzaakt door toxines en onvoldoende bescherming van hersencellen. Het genetische defect kan de productie van toxines bevorderen of resulteren in onvoldoende bescherming tegen toxines.
Risicofactoren voor levensstijl
De ziekte van Huntington treedt meestal op op middelbare leeftijd en de juveniele vorm ontwikkelt zich nadat de normale neurologische ontwikkeling is begonnen.
In tegenstelling tot sommige erfelijke aandoeningen is er geen probleem met de vorming van de hersenen bij de ziekte van Huntington - in plaats daarvan is er een probleem metonderhoudengezondheid van de hersencellen nadat ze zich al voldoende hebben gevormd.
Er zijn enkele populaties met een iets hogere incidentie van de ziekte van Huntington, maar er zijn geen leefstijlfactoren of gewoonten waarvan is aangetoond dat ze de aandoening veroorzaken of helpen voorkomen.
De ziekte van Huntington komt over de hele wereld voor, met een iets lagere prevalentie in Aziatische landen dan in Europa, de VS en Australië. De aandoening komt ook iets vaker voor bij vrouwen dan bij mannen, en iets vaker voor bij mensen met een lagere leeftijd. sociaaleconomisch niveau.
Deskundigen zijn niet zeker over de reden voor deze trends, en momenteel wordt begrepen dat sommige populaties waarschijnlijk de oorzakelijke mutatie dragen.
Andere verklaringen voor de verschillende incidentie tussen verschillende populaties zijn onder meer:
- Onderzoekers suggereren dat genetische tests en identificatie van de ziekte kunnen verschillen tussen verschillende populaties en dat dit een reden zou kunnen zijn voor de variatie in diagnose, in plaats van een feitelijk verschil in het voorkomen ervan.
- Onderzoekers suggereren dat vrouwen mogelijk meer vatbaar zijn voor een hoger aantal CAG-herhalingen dan mannen.
- Onderzoek toont aan dat het hebben van cognitieve en motorische gebreken kan leiden tot lagere inkomensniveaus voor degenen die door de aandoening worden getroffen en hun nakomelingen.
Een woord van Verywell
De ziekte van Huntington wordt veroorzaakt door een erfelijk genetisch defect op chromosoom vier. Het fysiologische proces waardoor het genetisch defect de effecten van de ziekte veroorzaakt, is complex en omvat progressieve schade aan bepaalde delen van de hersenen.
Hoewel er niets kan worden gedaan om de ontwikkeling van de ziekte van Huntington te voorkomen of om de neurologische degeneratie om te keren als u het veroorzakende gen heeft geërfd, kan een begrip van de biologische oorzaak uiteindelijk leiden tot ontdekkingen die de progressie van de ziekte kunnen helpen voorkomen bij mensen met de genmutatie.